Research
NHGRI GREAT Scholar | UC Santa Cruz Genomics Institute
Through the NIH NHGRI-funded GREAT Scholars program, I am paired with Dr. Benedict Paten's lab at the UC Santa Cruz Genomics Institute, working on performance improvements to pangenome graph tooling in the vg toolkit.
Porting vg-anchors to C++
My project is centered on vg-anchors, a tool in the pangenome-guided assembly (PGA) pipeline that is part of the vg toolkit. The PGA pipeline takes a pangenome graph and a sample's long sequencing reads and produces a phased local genome assembly, meaning it reconstructs which copy of a chromosome came from which parent, even in structurally complex regions.
A key step in this pipeline is anchor generation. Anchors are short, potentially haplotype-informative k-mers derived from unique paths within leaf snarls (sites of variation in the graph that contain no nested sub-variants). Because reads that support the same anchor likely originate from the same haplotype, anchors are used to partition reads before assembly.
The existing anchor generation code is written in Python. My work is to port it to C++ and introduce algorithmic improvements to reduce runtime and memory use, making the tool practical for large-scale pangenomes, which are expected to grow to thousands of haplotypes in coming years.
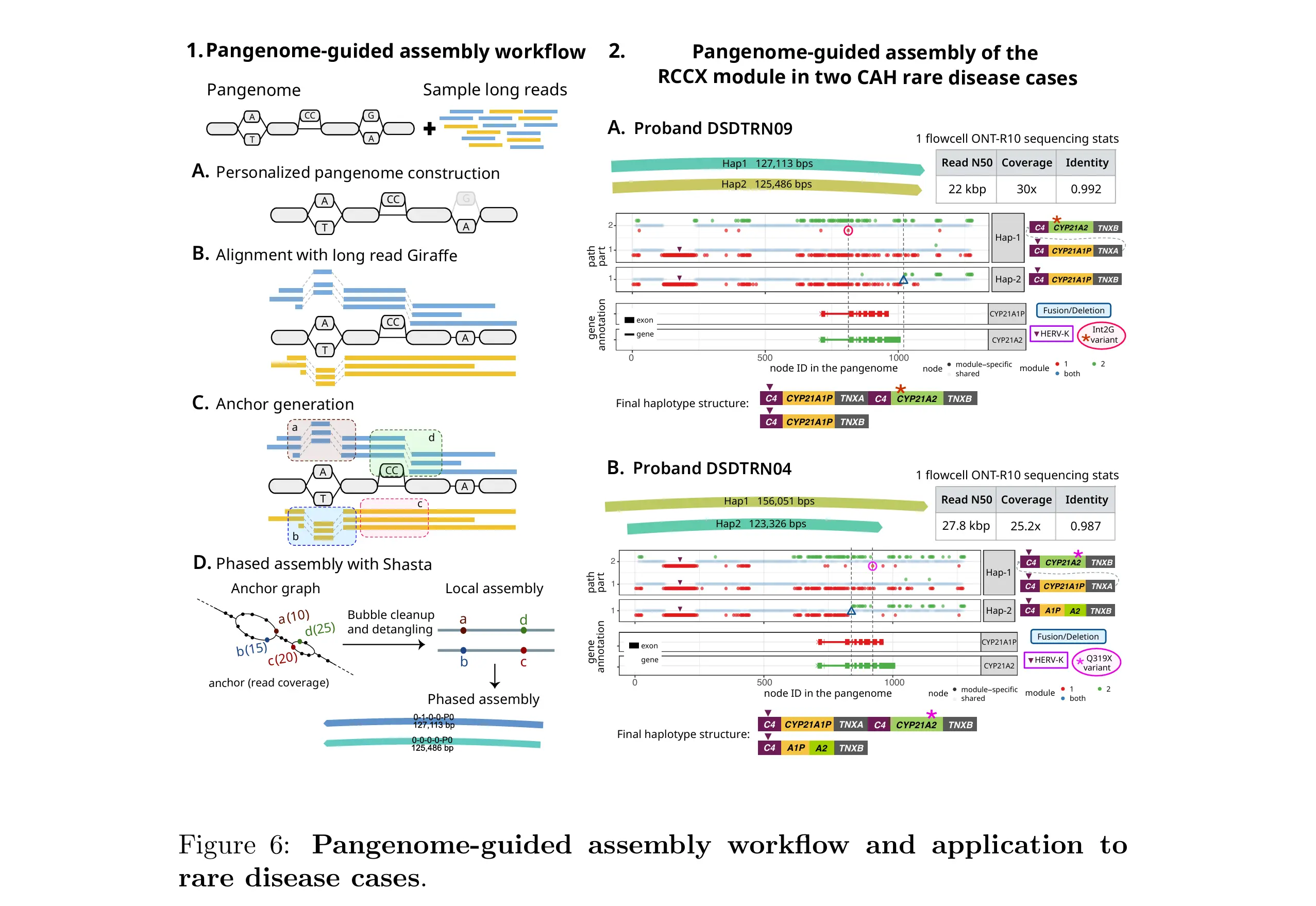
Background: Why Pangenomes?
Traditional genomics compares sequencing reads against a single linear reference genome, the GRCh38 human reference. This causes reference bias: variants that differ from the reference are harder to detect, especially in structurally complex regions like segmental duplications.
A pangenome graph encodes many haplotypes simultaneously. Each node in the graph holds a DNA sequence, and each path through the graph represents a known haplotype. The Human Pangenome Reference Consortium (HPRC) has released graphs built from hundreds of diverse human genomes, giving aligners a much richer reference to map reads against.
The vg toolkit's Giraffe aligner maps reads, both short Illumina reads and long Oxford Nanopore or PacBio HiFi reads, to these pangenome graphs efficiently. Personalized pangenomes take this further: before alignment, the graph is pruned down to haplotypes most compatible with the sample's own k-mer composition, reducing noise from variants the sample doesn't carry.
Current Status
This is my first research experience and it's still early. Right now I'm focused on understanding the existing Python implementation, the vg codebase, and the biology behind snarl-based anchor generation. I'm working toward producing a C++ implementation that passes the same correctness tests and measurably improves performance.
I'll update this page as the project develops.
Program
NIH NHGRI GREAT Scholars
California State University Monterey Bay
Host Lab
Computational
Genomics Lab
UC Santa Cruz Genomics Institute
PI: Dr. Benedict Paten